Familiärer Darmkrebs
Dickdarmkrebs (kolorektales Karzinom) zählt mit ca. 70.000 Neuerkrankungen pro Jahr und einem Lebenszeitrisiko von 4-6% zu den am häufigsten auftretenden bösartigen Tumorerkrankungen des höheren Lebensalters in Westeuropa. Karzinome entwickeln sich überwiegend aus zunächst gutartigen Schleimhautwucherungen (Polypen), die im Laufe der Zeit zu bösartigen Tumoren entarten können. Aber auch eine Tumorentstehung ohne vorausgehende Polypenbildung ist möglich. Die meisten Darmkrebserkrankungen sind sporadisch, d.h. nicht genetisch bedingt, und das Erkrankungsrisiko wird vordergründig durch das Lebensalter und Umwelteinflüsse beeinflusst.
Allerdings besteht in 20-30% der Fälle eine familiäre Häufung und bei ca. 5% lassen sich krankheitsverursachende genetische Veränderungen identifizieren, die zu einem erblichen Tumorprädispositionssyndrom führen. Betroffene von Tumorprädispositionssyndromen erkranken in der Regel früher und häufiger als die Allgemeinbevölkerung an bestimmten Tumoren. Neben Darmkrebs können dabei auch weitere Tumorarten auftreten. Aus diesem Grund existieren für diese Personen strengere Krebsfrüherkennungsempfehlungen. Da eine ursächliche genetische Veränderung zudem mit einer Wahrscheinlichkeit von bis zu 50% von Generation zu Generation weitervererbt wird, hat die Diagnose eines Tumorprädispositionssyndroms stets auch Bedeutung für Familienangehörige.
Auf den folgenden verlinkten Seiten möchten wir Ihnen weiterführende Informationen zu verschiedenen Tumorprädispositionssyndromen des Familiären Darmkrebses geben. In der Übersichtsgraphik (Abb. 1) wurden die verschiedenen Erkrankungen und die involvierten Signalwege innerhalb einer Zelle zusammengefasst. Detailliertere Informationen zu jeder Erkrankung finden Sie unter den entsprechend Links weiter unten auf dieser Seite.

- Abbildung 1: Tumorprädispositionssyndrome des Familiären Darmkrebses. Die verschiedenen Syndrome sind in gelb hervorgehoben, und die betroffenen Proteine wurden in blau markiert. (Modifiziert nach Short E und Sampson J., 2019, doi: 10.1016/bs.adgen.2018.11.002)
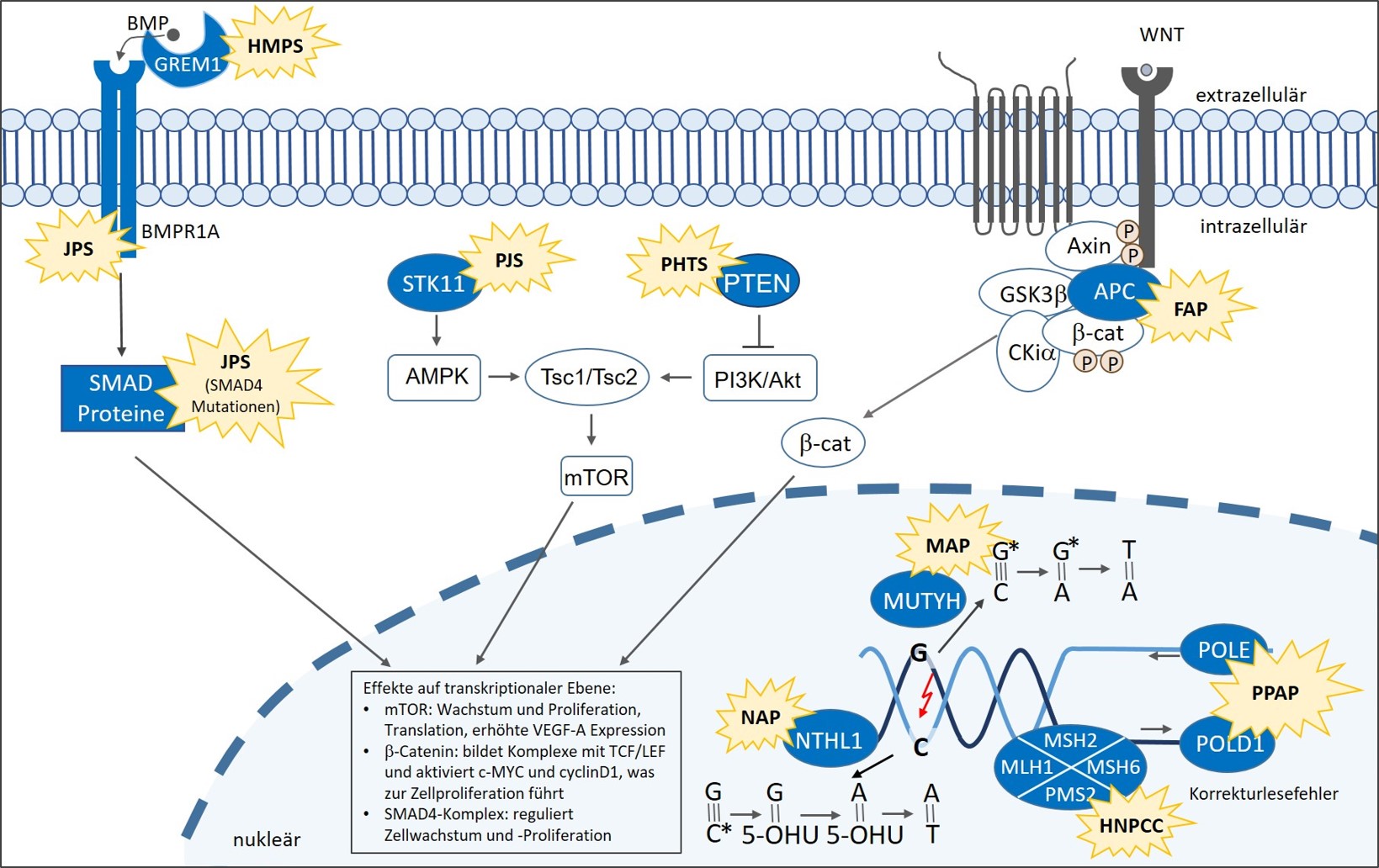
Eines der häufigsten Tumorprädispositionssyndrome ist das Lynch Syndrom (früher HNPCC), das ohne eine gesteigerte Polypenbildung einhergeht.
Andere Syndrome basieren auf der der vermehrten Bildung von Darmpolypen, die aufgrund von Veränderungen in den Zellen der Darmschleimhaut entstehen. Sie sind bei einer Darmspiegelung als wenige Millimeter bis einige Zentimeter große Ausstülpungen aus der Darminnenwand sichtbar. Ihre Form ist sehr unterschiedlich: Das Spektrum reicht von flachen, kaum sichtbaren Erhebungen über kleine "Knubbel" bis zu gestielten Geschwülsten, die weit in den Darm hineinragen. Die gesteigerte Polypenbildung wird als "Polypose" bezeichnet. Einzelne Dickdarmpolypen treten in der Allgemeinbevölkerung mit zunehmendem Alter häufig auf und sind oft symptomlos, stellen jedoch Krebsvorstufen dar. In Deutschland wird allen Personen ab dem 50. Lebensjahr die regelmäßige Früherkennung mittels Darmspiegelung empfohlen, weil Darmpolypen bei der Untersuchung erkannt und entfernt werden können, so dass eine Darmkrebserkrankung im besten Fall vermieden wird. Am häufigsten kommen adenomatöse (tubuläre oder tubulovillöse) und serratierte (bzw. hyperplastische) Polypen vor, sehr selten dagegen juvenile oder hamartomatöse Läsionen. Die Wahrscheinlichkeit für eine genetische Ursache steigt mit der Anzahl der Polypen. Weiterhin treten bei erblichen Polyposen gehäuft auch Polypen im oberen Magen-Darm-Trakt oder Tumore in weiteren Organen auf. Eine genetische Abklärung auf das Vorliegen einer erblichen Polypose sollte ab einer Anzahl von ≥10 Dickdarmpolypen unabhängig vom Typ oder ≥ 5 serratierten Polypen oder ≥ 2 hamartomatösen (PJS-Typ, juvenil) Polypen eingeleitet werden. Unabhängig von der Anzahl kann auch ein ungewöhnlich junges Alter, bei Erstauftreten oder der Nachweis von Polypen im oberen Gastrointestinaltrakt (Magen, Dünndarm) Anlass zur genetischen Abklärung geben.
Zu den erblichen Polyposen zählen:
- die Familiäre Adenomatöse Polyposis (FAP)
- die MUTYH-assoziierte Polyposis (MAP)
- das NTHL1-assoziierte Polypose (NAP)
- die Polymerase Proofreading-assoziierte Polyposis (PPAP)
- das Peutz-Jeghers-Syndrom (PJS)
- das PTEN-Hamartom-Tumor-Syndrom (PHTS)
- die Familiäre juvenile Polyposis (JPS)
Ob auch das Serratierte Polyposis Syndrom (SPS), die häufigste Polypose, zu den erblichen Erkrankungen zählt, ist Gegenstand aktueller Forschung. Da eine sichere Diagnosestellung allein anhand des klinischen Bildes oft nicht möglich und der Umfang der erforderlichen Früherkennungsmaßnahmen je nach betroffenem Gen variabel ist, kommt der molekulargenetischen Untersuchung aller in Frage kommender Gene ein hoher Stellenwert bei der Diagnostik zu.
Das Erbliche Gemischte Polyposis Syndrom (HMPS - Hereditary Mixed Polyposis Syndrome) konnte bislang nur bei der Ashkenazi Bevölkerungsgruppe nachgewiesen werden und scheint auf einen einzelnen Vorfahren zurückzuführen zu sein. Es resultiert aus einer Keimbahnmutation in dem BMP-Signalweg, nämlich einer spezifischen Überexpression der Sekretion von GREM1. Einzigartig bei diesem Syndrom ist das Fehlen bekannter extrakolonischer Manifestationen.
Als Partner des Deutschen Konsortiums Familiärer Darmkrebs bieten wir Ihnen eine genetische Beratung und Diagnostik bei Verdacht auf familiären Darmkrebs an. Wir möchten im Rahmen des Deutschen Konsortiums Familiärer Darmkrebs unsere Patienten und ihre Familien unterstützen, fachliche Standards setzen, die wissenschaftliche Forschung bei erblichen Krebserkrankungen fördern und die behandelnden Ärzte umfassend informieren.