MUTYH-assoziierte Polyposis (MAP)
Die MUTYH-assoziierte Polyposis (MAP, Häufigkeit ca. 1:20.000 bis 1:60.000) ist charakterisiert durch das Auftreten von dutzenden bis hunderten adenomatöser Polypen des Dickdarms, die mit hoher Wahrscheinlichkeit im Laufe der Zeit zu bösartigen Tumoren (kolorektale Karzinome) entarten können. Weniger häufig treten auch sessile und hyperplastische Polypen auf. Das durchschnittliche Diagnosesalter beträgt 45 Jahre, mit einer asymptomatischen Polypenbildung muss allerdings bereits Jahre vorher gerechnet werden. Unbehandelt liegt das Lebenszeitrisiko an Darmkrebs zu erkranken bei ca. 70-80% und ist unter regelmäßiger koloskopischer Kontrolle und Entfernung aller Polypen deutlich geringer. Allerdings scheint es bei bis zu einem Drittel der Betroffenen zu keiner oder allenfalls sehr milder Polypenbildung vor Auftreten eines kolorektalen Karzinoms zu kommen. Darüber hinaus kommt es bei ca. 25% der Patienten auch im Zwölffingerdarm (Duodenum) zu Polypenbildung, das Lebenszeitrisiko für ein Duodenalkarzinom ist mit 4% ebenfalls leicht erhöht, weswegen auch eine endoskopische Überwachung des oberen Magen-Darm-Trakts sinnvoll ist. Sofern eine endoskopische Abtragung aller Polypen nicht mehr erreicht werden kann, oder bereits ein Karzinom aufgetreten ist, kann die operative Entfernung des Dickdarms notwendig werden. Auch nach (inkompletter) Entfernung des Dickdarms (Kolektomie) sollte der verbliebene Enddarmanteil regelmäßig auf neu entstandene Polypen kontrolliert werden.
Auch Tumore anderer Organe treten bei MAP insgesamt häufiger, jedoch nicht wesentlich früher auf als in der Allgemeinbevölkerung (Lebenszeitrisiko ca. 38%). Insbesondere scheint ein moderat erhöhtes Risiko für maligne Tumore der Ovarien (Lebenszeitrisiko ca. 6%-14%), der Harnblase (Frauen ca. 6%-8%, Männer ca. 6%-25%), der Gebärmutter (ca. 3%) und der Haut (17%) zu bestehen. Verlässliche Daten hinsichtlich weiterer Tumorarten (bspw. der Schilddrüse, der Bauchspeicheldrüse oder der Brust) sind derzeit nicht verfügbar oder widersprüchlich. Weiterhin werden gutartige Drüsenkörperzysten des Magens (11%), gutartige Nebennierentumore und Knochenzysten des Unterkiefers bei MAP beobachtet.
VERERBUNG
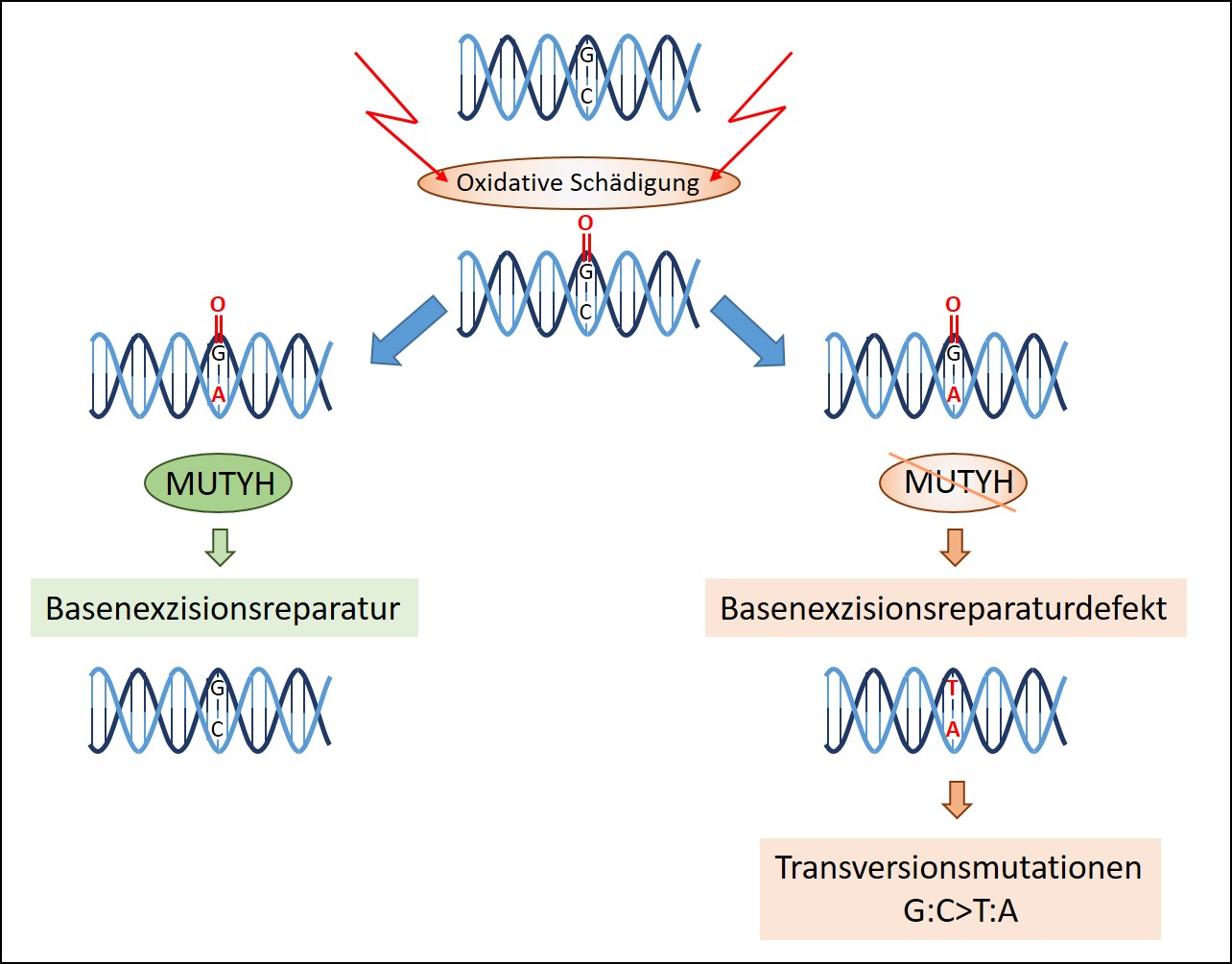
Die MAP wird durch biallelische Keimbahnmutationen (Veränderungen sowohl der mütterlichen als auch der väterlichen Genkopie) des MUTYH-Gens verursacht. Die Genveränderungen werden in aller Regel von beiden Eltern geerbt. MUTYH ist normalerweise an der Reparatur von oxidativen DNA-Schäden beteiligt, die durch Zellalterungsprozesse und Umwelteinflüsse hervorgerufen werden. Der Funktionsverlust des Gens führt zu einer genetischen Instabilität in einzelnen Zellen und in der Folge zu einer beschleunigten Tumorentstehung (Abbildung 1).

- Abbildung 1: Die Rolle von MUTYH bei der Basenexzisionsreparatur und der somatischen Signatur. Defektes MUTYH bei Darmkrebs führt zu einer Anreicherung für Transversionsmutationen.
Die MUTYH-assoziierte Polyposis folgt einem autosomal-rezessiven Erbgang, dies bedeutet, dass Nachkommen von MAP-Betroffenen in jedem Fall eine der beiden Mutationen im MUTYH-Gen erben und Anlageträger*innen bezeichnet werden. Diese erkranken nicht selbst, da vom nicht betroffenen Elternteil in der Regel eine zweite, gesunde Genkopie geerbt wird. Ein Erkrankungsrisiko für die Nachkommen von MAP-Betroffenen besteht nur dann, wenn das nicht betroffene Elternteil zufällig ebenfalls Anlageträger*in ist. Die Wahrscheinlichkeit in der Allgemeinbevölkerung beträgt hierfür ca. 1,5% (Heterozygotenfrequenz). Jedes Kind erbt in diesem Fall mit einer Wahrscheinlichkeit von 50% auch vom anderen Elternteil eine mutierte Genkopie und kann auch nur dann erkranken. Geschwister von MAP-Betroffenen tragen mit einer Wahrscheinlichkeit von 25% ebenfalls zwei mutierte Genkopien und mit einer Wahrscheinlichkeit von 50% nur eine mutierte Genkopie. Insbesondere für Geschwister (Risikopersonen) besteht die Möglichkeit einer prädiktiven (vorhersagenden) Testung ab dem 18. Lebensjahr, bei der die Veränderungen entweder nachgewiesen oder ausgeschlossen werden.
FRÜHERKENNUNG
Biallelische Mutationsträger*innen und deren Geschwister, bei denen die ursächlichen Genveränderungen noch nicht ausgeschlossen wurden, sollten lebenslang die folgenden Früherkennungsuntersuchungen wahrnehmen:
- komplette Koloskopie ab. 18.-20. Lebensjahr mit Entfernung aller Polypen > 5 mm 1- bis 2-jährlich, je nach Polypenlast
- Ösophagogastroduodenoskopie ab 25.-30. Lebensjahr mindestens 3-jährlich, bei Duodenalpolypen häufiger entspr. SPIGELMAN-Klassifikation
- Kolektomie bei endoskopisch nicht beherrschbarer Polypenlast, gleiche Kontrollintervalle postoperativ
Verwandte von MAP-Betroffenen, die nur eine mutierte Genkopie tragen (heterozygote Anlageträger) sollten wegen des gering erhöhten Darmkrebsrisikos erstmalig 10 Jahre vor dem Erkrankungsalter der Indexperson in der Familie, spätestens im 40.-45. Lebensjahr koloskopiert werden.
LITERATUR / LEITFADEN / SELBSTHILFEGRUPPE
LITERATUR
MUTYH Polyposis. Nielsen M., InfanteE., Brand R. Genereviews