Von-Hippel-Lindau-Syndrom
Das Von-Hippel-Lindau-Sydrom (VHL-Syndrom) ist eine seltene erbliche neurokutaneTumorerkrankung (Prävalenz 1:50.000), welche mit der Entstehung von meist gutartigen Tumoren einhergeht. Tumoren der Von-Hippel-Lindau-Krankheit entwickeln sich meistens im Gehirn und in der Netzhaut der Augen. In anderen Organen können sich andere Tumorarten entwickeln, darunter Tumoren in den Nebennieren (Phäochromozytome) und Zysten in den Nieren, der Leber oder der Bauchspeicheldrüse. Bei den betroffenen Menschen nimmt das Risiko für Nierenkrebs mit zunehmendem Alter zu.
Phänotypisch kann zwischen VHL-Typ I und VHL-Typ II unterschieden werden: VHL-Typ I ist charakterisiert durch das Auftreten von Hämangiomen in der Netzhaut und/oder im Zentralnervensystem, Nierenzellkarzinomen und/oder neuroendokrinen Tumoren. Das Risiko für Phäochromozytome ist allerdings sehr gering. VHL-Typ I ist mit nonsense-Mutationen oder Deletionen größerer Genabschnitte assoziiert. Im Gegensatz dazu ist bei VHL-Typ II das Risiko für die Entstehung von Phäochromozytomen sehr hoch, und häufig werden bei Betroffenen missense-Mutationen in VHL detektiert.
Die Symptome hängen von der Größe und der Lage der Tumoren ab. Kinder können unter Kopfschmerzen leiden und sich schwindlig oder schwach fühlen. Dazu können Sehstörungen, die bei wachsenden Tumoren der Netzhaut auch zu Sehverlust führen können, und Bluthochdruck kommen. Es kann zu einem Koordinationsverlust kommen. Etwa 10 Prozent der betroffenen Kinder hat einen Tumor des Innenohrs, was das Gehör beeinträchtigen kann. Ohne Behandlung können die Betroffenen erblinden, Hirnschäden erleiden oder sterben. Todesfälle sind normalerweise die Folge von Komplikationen der Gehirnangiome oder des Nierenkrebses.
Erkrankung | OMIM-P | ORPHA | Gen | OMIM-G | Vererbung | Prävalenz | Manifestation |
|---|---|---|---|---|---|---|---|
von Hippel-Lindau Syndrom | 193300 | VHL CCND 1 | autosomal dominant | 1:50.000 | Kindheit - Erwachsener |
INDIKATION
- Zwei oder mehr Hämangioblastome des Gehirns oder der Retina oder
- ein Hämangioblastom in Kombination mit einer viszeralen Manifestation wie Nieren- / Pankreaszysten, Nierentumor, Phäochromozytom und seltener Felsenbeintumor, papilläres Zystadenom und neuroendokriner Tumor des Pankreas.
Bei positiver Familienanamnese bei Patient*innen mit mindestens einem der folgenden Befunde:
- Angiom der Retina
- Hämangioblastom des Gehirns oder Rückenmarks
- Phäochromozytom
- Multiple Zysten des Pankreas oder der Niere oder
- Nierenzellkarzinom vor dem 60. Lebensjahr
FRÜHERKENNUNG
Altersunabhängig sollte jede Person mit Von-Hippel-Lindau-Syndrom eine jährliche klinische Untersuchung wahrnehmen. Bestandteile dieser ärztlichen Vorstellung sind Blutdruckkontrollen, neurologische Untersuchungen sowie die Evaluation von Seh- oder Hörverschlechterungen. Darüber hinaus sollten Personen mit VHL über Symptome und Zeichen der Erkrankung informiert werden. Idealerweise werden alle VHL-Patienten von einem mit dem Von-Hippel-Lindau-Syndrom vertrauten Arzt betreut, der auf ein multidisziplinäres Team zurückgreifen kann.
Hinsichtlich der einzelnen Manifestationen im Rahmen eines VHL schlägt die American Association for Cancer Research (AACR) folgende Früherkennungsempfehlungen vor:
Retinale Hämangioblastome
- Jährlich augenärztliche Untersuchung inklusive Retina-Diagnostik ab Geburt
Phäochromozytome
- Blutdruckkontrollen bei jeder ärztlichen Vorstellung ab 2 Jahren
- Jährlich freie Metanephrine im Plasma (PFM) oder fraktionierte Metanephrine im 24-Stunden-Urin ab 2 Jahren:
- Wenn PFM ≥4x oberhalb des Referenzwertes: Vereinbar mit PHEO, Bildgebung zur Lokalisation sollte angeschlossen werden
- Wenn PFM 2x-4x oberhalb des Referenzwertes: Wiederholung der Untersuchung in 2 Monaten
- Wenn PFM marginal erhöht: Wiederholung der Untersuchung in 6 Monaten (oder Clonidin-Suppressionstest, zum Ausschluss falsch positiver Werte)
Endolymphatische Sack Tumore
- Alle zwei Jahre Audiogramm ab 5 Jahren
ZNS-Hämangioblastome
- Alle zwei Jahre (ab dem Erwachsenenalter ggf. jährlich) MRT-Schädel mit und ohne Kontrastmittel + Dünnschichtaufnahmen des inneren Gehörgangs ab 8 Jahren
Nierenzellkarzinome
- Jährlich MRT-Abdomen ab 10 Jahren (kann zusammen mit dem Screening für pankreatische NET erfolgen)
Pankreatische neuroendokrine Tumore
- Jährlich MRT-Abdomen ab 10 Jahren (kann zusammen mit dem Screening für Nierenzellkarzinome erfolgen)
GENETIK
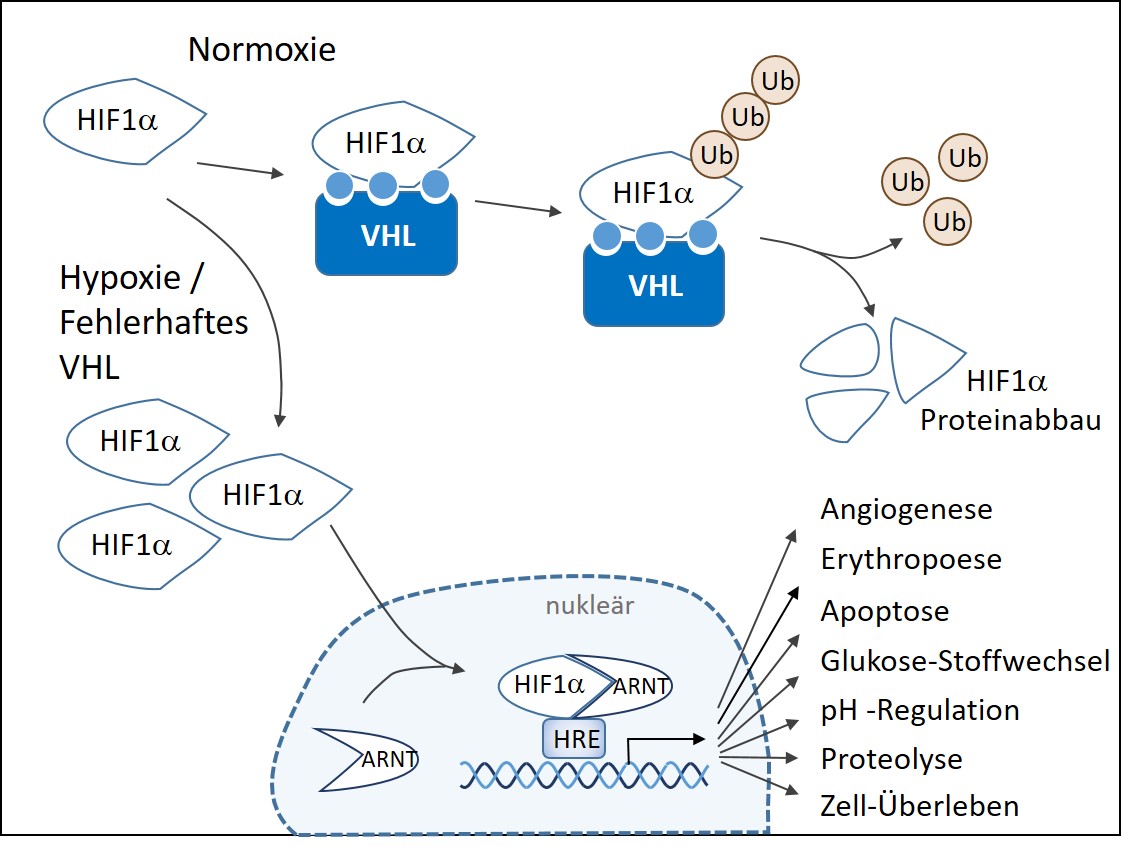
Ursächlich für das von-Hippel-Lindau-Syndrom sind Mutationen im Tumorsuppressorgen VHL. Das VHL-Gen ist auf Chromosom 3 lokalisiert und kodiert das VHL-Protein (pVHL). Verschiedene Funktionen des VHL-Proteins sind bis heute aufgeklärt worden, jedoch ist die bis heute am besten verstandene Funktion die Regulation anderer Gene als Antwort auf die Sauerstoff-Konzentration in der Zelle. Bei normalem Sauerstoffgehalt (Normoxie) wird HIF-1α in der Zelle rasch abgebaut. Das VHL-Protein bildet mit HIF-1α und anderen Proteinen einen Komplex, der zum sauerstoffabhängigen Proteinabbau von HIF-1α führt (Abb.1).
Ist der Sauerstoffgehalt niedrig (Hypoxie) kann HIF-1α nicht mehr an VHL binden und akkumuliert in der Zelle. Im Zellkern assoziiert es mit dem Protein ARNT ("arylhydrocarbon receptor nuclear translocator") und reguliert die Expression von hypoxieregulierten (HRE) Genen über die Bindung an spezifische DNA-Sequenzen. Eine hohe HIF-α Konzentration signalisiert der Zelle zum Beispiel, mehr Botenstoffe zur Bildung neuer Blutgefäße zu generieren, um durch neue Gefäße eine verbesserte Sauerstoffversorgung zu gewährleisten. Da die Versorgung mit Blutgefäßen aber auch das Tumorwachstum begünstigen kann, ist die intrazelluläre Konzentration des kurzlebigen HIF-α Proteins eine sehr genau regulierte Stellgröße. Eine Vielzahl der durch HIF1a regulierten Gene ist zum heutigen Zeitpunkt bekannt. Dazu gehören u.a. VEGF (vascular endothelial growth factor), EPO (erythropoietin), GLUT-1 (glucose transporter 1), PDGF (platelet derived growth factor), TGF α (transforming growth factor alpha) und viele andere.
Das „Notsignal Sauerstoffmangel“ kann aber auch durch Mutationen im VHL-Gen nachgeahmt werden, die den Bauplan des VHL-Proteins derart stören, dass das veränderte VHLProtein die Bindung von HIF-α nicht oder nur noch eingeschränkt bewerkstelligen kann. Im schlimmsten Fall kann es ganz fehlen. In der Folge wird HIF-α nicht mehr ausreichend entsorgt, und es kommt zur Tumorbildung mit verstärkter Blutgefäßbildung
Keimbahnmutationen in VHL führen nicht unmittelbar zur Entartung. Erst nach Ausfall der zweiten, intakten VHL-Kopie durch spontane somatische Mutationen kann HIF-1α nicht mehr an pVHL binden, wodurch es in der Zelle akkumuliert und Zellproliferation und Neovaskularisierung von Tumoren verursachen kann.

- Abbildung 1: Schematische Darstellung der sauerstoffabhängigen Regulierung des Hypoxie-induzierbaren Faktors HIF-1a. Unter Normoxie Bedingungen wird HIF-α chemisch so modifiziert (blaue Kreise), dass es an den pVHL Komplex binden kann. Nach der Polyubiquitinierung wird HIF-α durch das Proteasom abgebaut. Ist der Sauerstoffgehalt der Zelle zu gering, erfolgt keine chemische Veränderung von HIF-α und seine Konzentration steigt in der Zelle an. In der Folge kommt es zu einer Kaskade von Wechselwirkungen über sogenannte HRE (hypoxia responsive elements) und zur Regulation sauerstoffkonzentrationsabhängiger Zielgene.
Das VHL-Syndrom folgt einem autosomal dominanten Erbgang, ca. 80 % der Patienten mit VHL haben ein betroffenes Elternteil und ca. 20 % haben VHL aufgrund von de novo Mutationen. Keimbahnmosaike bei gesunden Eltern sind ebenfalls beschrieben. Bei gesicherter klinischer VHL-Diagnose werden in mehr als 98 % der Patienten Mutationen in VHL gefunden (ca. 72 % Punktmutationen und 28 % partielle bzw. komplette Deletionen). Die Penetranz von VHL-Mutationen ist sehr hoch, fast alle Individuen mit Keimbahn VHL-Mutationen entwickeln Symptome bis zum 65 LJ. Die Inzidenz wird mit ca. 1 : 35 000 Lebendgeburten angegeben.
LITERATUR / LEITLINIE / SELBSTHILFEGRUPPE
LITERATUR
Von Hippel-Lindau Syndrome. van Leeuwaarde RS, Ahmad S, Links TP, Giles RH. GeneReviews
Von-Hippel-Lindau-Erkrankung: Aktuelle Zusammenstellung. Heller R. 2011, Dtsch Arztebl 108: A-2105
SELBSTHILFEGRUPPE