Neurofibromatose Typ 2
Die Neurofibromatose Typ II (NF II), die auch als zentrale Neurofibromatose bezeichnet wird, ist eine erbliche Tumorerkrankung. Ihr Hauptmerkmal ist das Vorkommen von gutartigen Hirntumoren, die sich symmetrisch im Bereich beider Hör- und Gleichgewichtsnerven entwickeln (vestibuläre Schwannome - VS) und zu Gleichgewichtsstörungen und schließlich zu völliger Taubheit führen. Schwannome an anderen Nerven im Kopf und in der Wirbelsäule sowie an anderen Stellen des Körpers können zum Verlust von Muskelfunktionen und Empfindungen führen, so dass viele NF2-Betroffene schließlich im Rollstuhl sitzen. Der zweithäufigste Tumor bei NF2 ist ein nicht-krebsartiger Tumor an der Auskleidung von Gehirn und Rückenmark, ein so genanntes Meningeom. Auch er kann zu Funktionsstörungen wie Kopfschmerzen und Krampfanfällen führen. Ein Tumor im Rückenmark, ein so genanntes Ependymom, betrifft etwa 20-40 % der Betroffenen, ist aber in der Regel nicht progressiv. Die meisten Patienten mit dieser Erkrankung leiden auch an Veränderungen der Augen. Ursache der NF II sind Mutationen eines Gens, das vermutlich Einfluss auf Form und Wanderungsverhalten bestimmter Zelltypen nimmt. Da die NF II genetisch bedingt ist, ist eine Heilung nicht möglich. Die Behandlung besteht in der Entfernung von Tumoren im Bereich des Gehirns und Rückenmarkes, sowie operativer Eingriffe im Bereich der Augen und der betroffenen Hirnnerven. Darüber hinaus zeigte sich, dass man mit einigen chemotherapeutischen Wirkstoffen (z.B. Bevacizumab, Lapatinib) das Tumorwachstum stoppen oder zumindest verlangsamen kann.
KLINISCHE BESCHREIBUNG
Betroffene Personen entwickeln unweigerlich Schwannome, die typischerweise beide Vestibularnerven betreffen und zu Hörverlust und Taubheit führen. Die Mehrheit der Patienten weist einen Hörverlust auf, der zu Beginn in der Regel einseitig ist und mit Tinnitus einhergehen oder ihm vorausgehen kann. Vestibuläre Schwannome können als erstes Symptom auch Schwindel oder Unausgeglichenheit verursachen. Übelkeit, Erbrechen oder echter Schwindel sind seltene Symptome, außer im Spätstadium der Erkrankung. Die anderen Haupttumoren sind Schwannome der anderen kranialen, spinalen und peripheren Nerven; Meningiome sowohl intrakranielle (einschliesslich Sehnervenmeningiome) als auch intraspinale und einige geringgradige Malignome des Zentralnervensystems (Ependymome). Ophthalmische Merkmale sind ebenfalls ausgeprägt und umfassen eine reduzierte Sehschärfe und Katarakt. Etwa 70% der NF2-Patienten haben Hauttumore (intrakutane plaqueähnliche Läsionen oder tiefer liegende subkutane knötchenförmige Tumore).
Als diagnostische Kriterien für eine Erkrankung bezeichnet man die Symptome, bei deren Vorliegen die klinische Diagnose gestellt werden darf. Für das Vorliegen einer definitiven NF II sind dies:
- Der Nachweis von bilateralen Akustikusneurinomen mittels bildgebender Verfahren.
- Ein Verwandter ersten Grades mit einer NF II und der Nachweis von Neurofibromen, Meningeomen, Gliomen, Schwannomen.
- Ein Verwandter ersten Grades mit einer NF II und der Nachweis einer juvenilen posterioren subcapsulären Katarakt (Linsentrübung im jugendlichen Alter).
- Zwei oder mehr feingeweblich bestätigte Schwannome oder Meningeome und Genetik von mindestens zwei Tumoren, die einen Verlust der Heterozygotie (Verlust beider Allele) für das Chromosom 22 aufweisen und zwei unterschiedliche NF2-Mutationen im Tumormaterial haben. Wenn eine SMARCB1-Mutation vorliegt, definiert dies eine SMARCB1-assoziierte Schwannomatose.
- Ein neuropathologisch bestätigtes Schwannom oder Meningeom und eine in der Blutbahn nachgewiesene SMARCB1-Mutation.
Folgende Kriterien machen das Vorliegen einer NF II wahrscheinlich:
- Einseitiges Akustikusneurinom vor dem 30. Lebensjahr und ein Meningeom, Schwannom, Gliom oder Linsentrübung.
- Mehrere Meningeome und ein Gliom, Linsentrübung oder Schwannom vor dem 30 Lebensjahr.
Vermutetet ein Arzt für Allgemeinmedizin das Vorliegen einer NF2, sollte der Patient zur weiteren Diagnostik an einen Spezialisten überwiesen werden (meist Neurologe). Als wichtigste diagnostische Kriterien gelten:
- Vorliegen eines Akustikusneurinoms auf einem Ohr und gleichzeitiges Vorliegen zwei oder mehr typischen Symptome, wie Katarakt und Hirntumoren, und es gibt einen Verwandten ersten Grades mit NF2.
- Vorliegen von Akustikusneurinomen auf beiden Ohren
Ein CT (Computertomographie) oder MRT (Magnetresonanztomographie) Scan des Gehirns können das Vorliegen eines Akustikusneurinoms oder anderen Gehirn- oder Rückenmarkstumoren bestätigen.
Eine Blutprobe wird entnommen und in ein Labor, das fehlerhafte Gen erkennen. Kann das fehlerhafte Gen identifiziert werden, gilt die Diagnose ebenfalls als einwandfrei bestätigt.
Eine regelmäßige Verlaufskontrolle der NF2 ist aufgrund der Tatsache, dass es eine kausale Therapie der NF nicht gibt, die wichtigste Möglichkeit zur Früherkennung neuer Symptome.
GENETIK
NF2 ist eine vererbbare genetische Erkrankung, die durch einen Fehler in einer Kopie des NF2-Gens auf Chromosom 22 verursacht wird und einem autosomal dominanten Erbgang folgt.
NF2 wird in etwa 40 % der Fälle von einem betroffenen Elternteil vererbt. In den übrigen Fällen beginnt die Veränderung des NF2-Gens bei dieser Person. Dies geschieht entweder in allen Zellen (65-70 %), was darauf hindeutet, dass die Veränderung bereits in der Ei- oder Samenzelle vorhanden war, aus der die erste Zelle bei der Empfängnis entstand, oder nur in einigen Zellen, was darauf hindeutet, dass der Gendefekt während der Entwicklung des Embryos auftrat (Mosaizismus genannt). Mosaik-NF2 ist milder, da nicht alle Zellen betroffen sind, und die Wahrscheinlichkeit, dass NF2 an Kinder weitergegeben wird, ist geringer als die üblichen 50 %.
Obwohl trunkierende Keimbahn-Mutationen (Nonsense und Frameshift) das häufigste Ereignis darstellen und einen schweren Verlauf verursachen, sind auch einfache und multiple Exon-Deletionen häufig. Eine Strategie zum Nachweis der letzteren ist für eine empfindliche Analyse von entscheidender Bedeutung.
Die Identifizierung des zugrundeliegenden Gendefekts ist besonders hilfreich bei der Beurteilung des potenziellen Schweregrads der Erkrankung, wobei man, wenn möglich, mit der Untersuchung des Tumors in isolierten Fällen statt des Bluts beginnt.
HINTERGRUNDINFORMATIONEN ZU MERLIN
Das krankheitsrelevante Gen kodiert für das Genprodukt Merlin, was ein Akronym für Moesin-Ezrin-Radixin like protein ist. Merlin ist in der Lage, auf verschiedene Signalwege der Zelle einen hemmenden Einfluss auszuüben und darüber die Proliferation und somit das Wachstum von Gewebe fein zu regulieren.
Die Tumorsuppressor-Aktivität von Merlin wurde zunächst mit einer Rolle bei der durch das Transmembranprotein CD44 vermittelten Kontaktinhibierung der Zellproliferation verbunden (Abb.1). Aktives Merlin interagiert mit dem Transmembranprotein CD44, welches häufig mit verschiedenen Wachstumsfaktorrezeptoren assoziiert ist, und inhibiert seine Aktivität. Durch die Modulierung der subzellulären Lokalisation des kleinen GTPase Proteins Rac1 beeinflusst Merlin die durch PAK (p21-activated kinase) vermittelte Signalgebung und passt die Organisation des Aktin-Zytoskeletts in Abhängigkeit von der Zelldichte an.

- Abbildung 1: Schematische Darstellung der Signalwege, die Merlin beeinflusst.
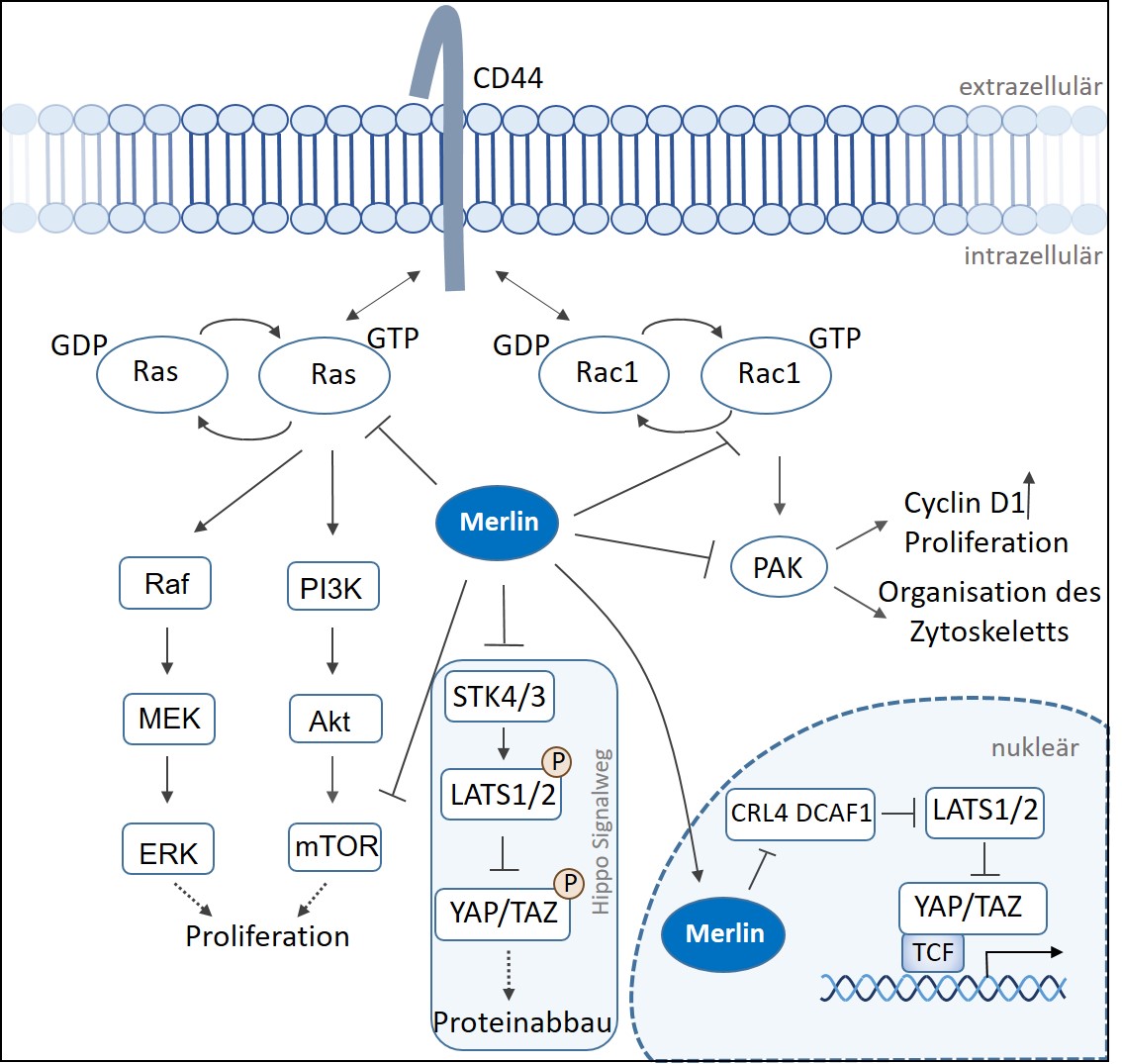
Merlin greift auch in den mTOR Signalweg ein, der an der Regulierung der Proteinsynthese, der Zellteilung, der Zellproliferation und der synaptische Plastizität von Zellen des Nervensystems und des Gehirns beteiligt ist. Der Verlust von Merlin führt zur Aktivierung des mTOR-Signalwegs und zur unkontrollierten Proliferation von Schwannzellen.
Weiterhin interagiert Merlin mit dem Hippo-Signalweg, der die Organgröße bei Tieren durch Regulierung der Zellproliferation und Zelltod steuert. Dabei wird über eine Signalkaskade das YAP/TAZ Protein im Zytoplasma phosphoryliert, wodurch seine Rolle als Transkriptionsfaktor geblockt und das Protein abgebaut wird. Im Zellkern selbst kann Merlin an die E3 Ubiqitin Ligase CRL4 DCAF1 binden und deren Aktivität inhibieren. Eine Transkription der Zielgene, die die Zellproliferation, das Überleben, die DNA Reparatur und die Genomintegrität durch Ubiquitinylierung von kritischen Regulatoren beeinflussen, wird unterbunden.
VORSORGE
Für Betroffene oder gefährdete Personen:
Jährliche MRT-Untersuchung, beginnend im Alter von etwa zehn bis 12 Jahren und mindestens bis zum vierten Lebensjahrzehnt; Hörprüfung, einschließlich BAER-Test; jährliche vollständige Augenuntersuchung.
Die Standardtherapie besteht zurzeit aus der maximalen Resektion der Meningeome, gefolgt von Bestrahlung der verbleibenden Krebszellen. Trotz verbesserter Operationsmethoden und Bestrahlungstherapien verbleibt dennoch eine Untergruppe aggressiv wachsender Meningeomen, die sich auf herkömmliche Weise nicht therapieren lassen. Hinzu kommt, dass nur sehr beschränkte alternative Behandlungsmethoden zur Verfügung stehen. So werden zum Beispiel in den Richtlinien, welche das US-amerikanische National Comprehensive Cancer Network (NCCN) 2011 veröffentlichte, lediglich drei Substanzen zur Behandlung von Meningeomen aufgeführt.
Zu vermeidende Wirkstoffe/Umstände:
Strahlentherapie von NF2-assoziierten Tumoren, insbesondere im Kindesalter, wenn das Malignitätsrisiko wahrscheinlich wesentlich höher ist.
Die frühzeitige Identifizierung von Verwandten, die die familienspezifische NF2-Pathogenvariante geerbt haben, ermöglicht eine angemessene Überwachung und damit eine frühere Erkennung und Behandlung von Krankheitsmanifestationen.
LITERATUR / LEITLINIE / SELBSTHILFEGRUPPE
LITERATUR
Neurofibromatosen. Dtsch Arztebl Int 2020; 117: 354-60; DOI: 10.3238/arztebl.2020.0354
Neurofibromatosis 2. D Gareth Evans, MD, FRCP. GeneReviews® [Internet].
SELBSTHILFEGRUPPE
Bundesverband Neurofibromatose (https://lfsa-deutschland.de/)