Familiäres atypisches multiples Muttermal- und Melanomsyndrom (FAMMM-PC)
Das familiäre atypische multiple Muttermal- und Melanom (FAMMM)-Syndrom ist eine genetisch bedingte Hauterkrankung (Genodermatose), die durch das Vorhandensein multipler melanozytärer Hautveränderungen (oft mehr als 50) und einer Familienanamnese von Melanomen gekennzeichnet ist und bei einer Untergruppe von Patienten ein erhöhtes Risiko für die Entwicklung von Bauchspeicheldrüsenkrebs und anderer Malignome nach sich zieht. Die Erkrankung ist durch Mutationen im CDKN2A-Gen bedingt.
Für Anlageträger besteht ein deutlich erhöhtes Risiko für Melanome (Hautkrebs) und ein Risiko von 17% für Bauchspeicheldrüsenkrebs. Auch Karzinome des Gastrointestinaltraktes und des Respirationstraktes sind im Zusammenhang mit CDKN2A-Mutationen beschrieben. Bei Nachweis einer krankheitsverursachenden Mutation liegt das Risiko einer Anlageträgerschaft für Nachkommen bei 50%. Für gesunde Familienangehörige besteht die Möglichkeit einer prädiktiven (vorhersagenden) Testung.
Erkrankung | OMIM-P | ORPHA | Gen | OMIM-G | Vererbung | Prävalenz | Manifestation |
|---|---|---|---|---|---|---|---|
FAMMM-PC | 155600, 606719 | CDKN2A | 600160 | autosomal dominant | unbekannt | alle Altersstufen |
KLINISCHER PHÄNOTYP
Der klinische Phänotyp des FAMMM-Syndroms zeigt eine große Heterogenität in Bezug auf das Vorhandensein von Nävi und die familiäre Prädisposition für Melanome und Pankreaskarzinome (hauptsächlich Adenokarzinome). Die Krankheit tritt am häufigsten bei Kindern und Jugendlichen auf, kann aber in jedem Alter auftreten. Das präsentierende Merkmal ist normalerweise eine hohe Gesamtkörper-Nävizahl (normalerweise mehr als 50). Die meisten sind klinisch typisch, aber einige können ein atypisches Aussehen haben (asymmetrisch, erhaben und/oder verschiedene Schattierungen von Braun, Braun, Schwarz oder Rot und oft von unterschiedlicher Größe), das einem frühen Melanom ähnelt und am häufigsten auf dem Rücken, der Brust , Gesäß, Brüste und Kopfhaut. Melanome können aus atypischen Muttermalen oder de novo entstehen und wurden bei einigen Patienten mit FAMMM-Syndrom bereits im zweiten bis dritten Lebensjahrzehnt berichtet. Menschen mit CDNK2A-Mutationen haben ein 90%iges Risiko, im Alter von 80 Jahren ein Melanom zu entwickeln, und ein um 20% erhöhtes Risiko, im Alter von 75 Jahren an Bauchspeicheldrüsenkrebs zu erkranken. Diese Mutationen sind auch mit einem jüngeren Erkrankungsalter verbunden. Andere Krebsarten, die selten mit dem FAMMM-Syndrom in Verbindung gebracht werden können, sind Brustkrebs, Speiseröhrenkrebs und Sarkome.
GENETIK
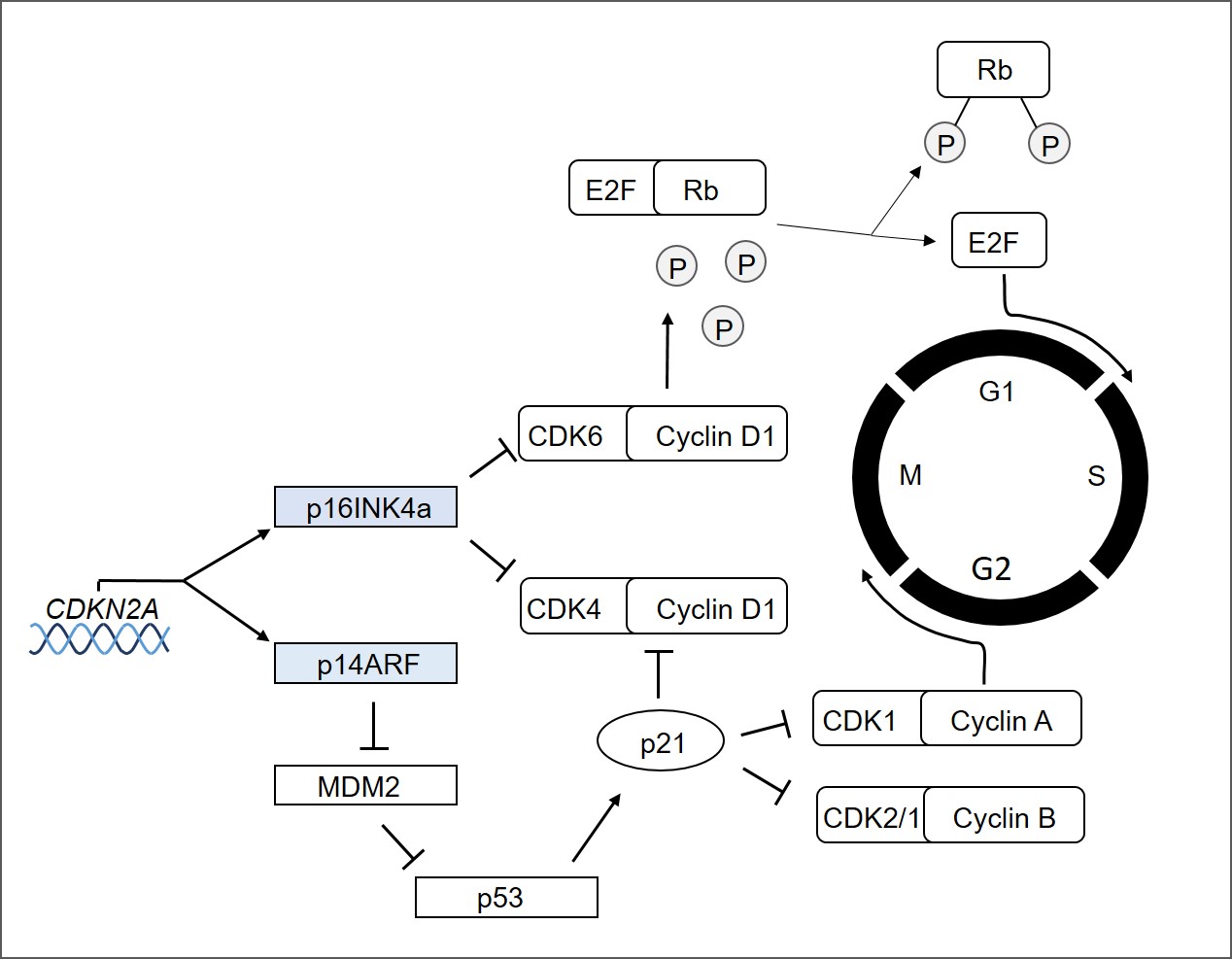
Das FAMMM-Syndrom wird mit Mutationen im 16p-Locus von CDKN2A in Verbindung gebracht. CDKN2A wird ubiquitär in vielen Geweben und Zelltypen exprimiert und kodiert für zwei Proteine, darunter das Mitglied der INK4-Familie p16 (oder p16INK4a) und p14ARF. Beide wirken als Tumorsuppressoren, indem sie den Zellzyklus regulieren (s. Abb. 1). p16INK4a hemmt die Cyclin-abhängigen Kinasen 4 und 6 (CDK4 und CDK6) und aktiviert dadurch die Retinoblastom (Rb)-Proteinfamilie, die den Übergang von der G1- zur S-Phase blockiert. Im Gegensatz dazu stabilisiert und aktiviert p14ARF das Tumorsuppressorgen p53 durch Hemmung von MDM2, wodurch der Abbau von p53 verhindert wird. Aktives p53 induziert die Expression von p21, einem negativen Zellzyklusregulator, der ein Inhibitor der CDK1-Cyclin A/B-Komplexe ist, wodurch das Fortschreiten von der G2-Phase zur Metaphase verhindert wird.
Somatische Mutationen von CDKN2A sind bei den meisten Krebserkrankungen beim Menschen verbreitet, wobei Schätzungen zufolge CDKN2A nach p53 das am zweithäufigsten inaktivierte Gen bei Krebs ist. Die Mutationen in den Proteinen p16INK4a und p14ARF führen zu einer Deregulation der Zellzyklusmaschinerie, die ein unkontrolliertes Wachstum der Zellen bedingt. Keimbahnmutationen von CDKN2A werden mit familiärem Melanom, Glioblastom und Bauchspeicheldrüsenkrebs in Verbindung gebracht. Das CDKN2A-Gen enthält auch einen von 27 single nucleotide polymorphisms (SNPs), die mit einem erhöhten Risiko für koronare Herzkrankheiten verbunden sind.
Das FAMMM-Syndrom wird autosomal-dominant mit unvollständiger Penetranz vererbt. Den Patienten und ihren Familien sollte eine genetische Beratung angeboten werden.

- Abb.1: Schema der Zellzyklus-Regulation durch CDKN2A. Das CDKN2A-Gen kodiert für zwei alternativ gespleißte Transkripte, p16INK4A und p14ARF, die sich in ihrem ersten Exon unterscheiden. Das p16INK4A-Protein hemmt die CDK4/6-Cyclin-D1-Komplexe, hält die Retinoblastom (Rb)-Proteine in einem dephosphorylierten Zustand und ermöglicht die Bindung und Inaktivierung der E2F-Transkriptionsfaktoren. Freies E2F sorgt für die Transkription verschiedener Proteine, von denen die meisten für den Übergang in die S-Phase notwendig sind. P16INK4A wird auch von E2F hochreguliert. p14ARF aktiviert den p53-Tumorsuppressor und verhindert durch die Expression von p21 das Fortschreiten des Zellzyklus' von G2 zu M.